Dans notre Lettre des Sciences[1] du 2 Septembre 2021, nous relations les performances remarquées de l’intelligence artificielle Alphafold développée par la filiale DeepMind de Google, dans le cadre du dernier concours CASP[2] de prédiction de structure 3D de protéines à partir de leurs seules séquences d’acides aminés.
Une des structures prédites par Alphafold a notamment permis de résoudre la conformation d’une protéine bactérienne impliquée dans des phénomènes de résistance aux antibiotiques et sur laquelle la communauté scientifique butait depuis près d’une décennie. Cette performance a été réalisée en seulement 30 minutes avec un degré de précision de 92%[3].
Récemment, Deepmind a publié le code source de son programme et annoncé qu’elle avait utilisé, entretemps, son IA pour prédire les formes de la quasi-totalité des protéines du corps humain, ainsi que les formes de centaines de milliers d’autres protéines trouvées dans 20 des organismes les plus étudiés, y compris la levure, les drosophiles et les souris.
Dans cet article, Inneance vous propose de plonger au cœur de cet IA qui fait tant parler, afin d’en éclaircir les mécanismes d’actions.
1. Alphafold : neurones artificiels et réseaux de neurones à convolution
Dans un article récent publié dans la prestigieuse revue scientifique Nature[4], DeepMind a notamment détaillé l’approche utilisée par son programme « Alphafold2 ». Initialement, le premier AlphaFold était constitué d’un réseau neuronal convolutif[5]. Cette typologie de réseau a permis de nombreuses percées en matière d’IA au cours de la dernière décennie, notamment dans les domaines de la reconnaissance d’images, reconnaissance des vidéos ou encore dans le traitement du langage naturel.
On peut se représenter un neurone artificiel comme une unité de calcul élaborée sur le modèle d’un neurone biologique. À l’instar de son homologue naturel, celui-ci est capable d’accepter un signal en entrée, de le modifier éventuellement et de le restituer à une ou plusieurs sorties. Ainsi, au sein d’un réseau de neurones artificiels, le traitement de l’information suit toujours la même séquence : les informations sont transmises sous la forme de signaux aux neurones de la couche d’entrée, où elles sont traitées. À chaque neurone est attribué un « poids » particulier (représentatif de la « force » de la connexion interneuronale) et donc une importance différente. Associé à une fonction dite d’activation, le poids permet de pondérer la valeur de sortie d’un neurone et ainsi décider quelles informations peuvent entrer dans le système. Réparti sur l’ensemble du réseau, ce mécanisme permet de connecter et d’activer un nombre plus ou moins grand de neurones. Les réseaux de neurones artificiels peuvent de fait être décrits comme des systèmes composés d’au moins deux couches de neurones – une couche d’entrée et une couche de sortie – et comprenant généralement des couches intermédiaires (Figure 1).
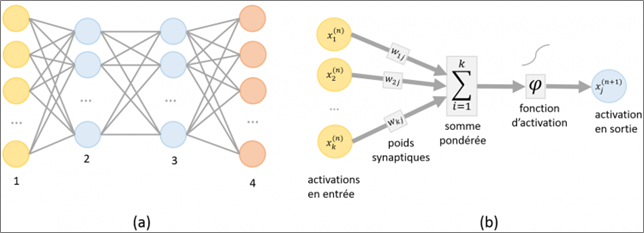
Figure 1 : Représentation schématique d’un réseau de neurones artificiels (a) et schéma d’activation (b) (Image source). (a) Représentation schématique d’un réseau de neurones artificiels à 4 couches. La couche d’entrée est représentée par la couche de neurones numéro 1 et la couche de sortie est représentée par la couche de neurones numéro 4. (b) Représentation schématique du mécanisme d’activation d’une couche de neurones. Chaque neurone de la couche possède un poids synaptique propre (wkj sur la figure). Lors de l’intégration de données, chaque neurone de la couche capte un signal d’entrée qui se combine au poids synaptique du neurone. La convergence et la somme de l’ensemble de ces informations associées à une fonction d’activation permettent de générer une donnée de sortie qui sera alors traitée par la couche de neurones suivante.
Dans le cadre du traitement d’informations complexes, les réseaux de neurones sont généralement structurés de telle sorte qu’il en existe plusieurs de distincts, chacun dédié au traitement d’une partie de l’information. Ces réseaux de neurones sont appelés des réseaux neuronaux convolutifs. La structuration de ces réseaux permet une segmentation du traitement de l’information qui permet, elle-même, in fine le traitement de l’ensemble de l’information (par exemple, le traitement d’images, de vidéos et de textes).
Traditionnellement, un réseau de neurones à convolution repose sur le principe mathématique du même nom. Par exemple, dans le cadre de l’analyse et du traitement d’une image, chaque zone de l’image est découpée en petites zones afin que chacune d’entre elles puisse être traitée par un neurone artificiel (unité de calcul). Chaque neurone appartenant à une même couche possède les mêmes paramètres de réglages. Cette strate de neurones possédant les mêmes paramètres est appelée « noyau de convolution ».
Un noyau de convolution permet d’analyser une caractéristique de l’image d’entrée. Pour analyser plusieurs caractéristiques, il est nécessaire d’empiler des noyaux de convolution indépendants, chaque noyau analysant une caractéristique de l’image. L’ensemble des strates ainsi empilées forme la « couche de traitement convolutif »[6].
Comparés à d’autres algorithmes de classification, les réseaux de neurones convolutifs utilisent relativement peu de prétraitement. Cela signifie que le réseau est seul responsable de l’évolution de ses propres filtres (apprentissage sans supervision), ce qui n’est pas le cas d’autres algorithmes plus traditionnels. Ainsi, l’absence de paramétrage initial et d’intervention humaine permet une capacité d’apprentissage automatique et évolutive, ce qui est un atout majeur des réseaux de neurones convolutifs. Cet avantage des réseaux de neurones convolutifs pourrait expliquer en partie pourquoi Deepmind s’est attaché dans un premier temps à cette typologie de réseau afin de développer son IA.
2. AlphaFold2 : CNN VS GNN
Cependant, lors du développement de la deuxième version d’AlphaFold, les équipes de DeepMind ont délaissé l’approche par convolution et mis en place une approche par réseau de neurones en graphiques (GNN pour Graph Neural Network), combinée aux mécanismes dit « d’attention ».
Les GNN sont une nouvelle forme de réseaux de neurones qui se basent sur des objets mathématiques dénommés « graphes ». En mathématique, un graphe est une structure de données se composant de plusieurs points (ou nœuds) reliés entre eux par des segments (ou bords). Par exemple, un graphe peut représenter un réseau social : les nœuds sont les utilisateurs et les segments représentent les relations entre eux. Au sein d’un graphe, les relations sont aussi importantes que les données elles-mêmes. Ainsi, chaque nœud et chaque segment peut posséder un ou plusieurs attributs[7]. Dans un contexte biologique, une molécule telle qu’une protéine repliée peut être pensée comme un graphe spatial où les acides aminés sont représentés comme un ensemble de nœuds et de segments reliés les uns aux autres (Figure 2).
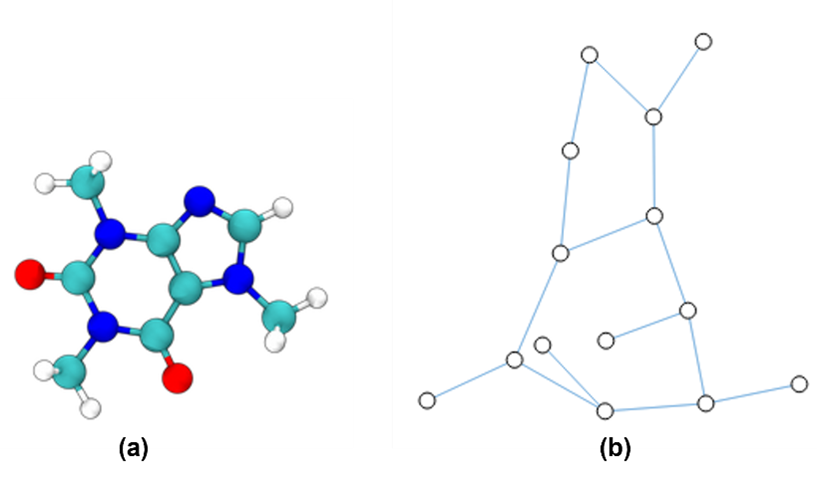
Figure 2 : (a) représentation en trois dimensions de la molécule de caféine, (b) représentation en graph de la molécule. Au sein d’une molécule, différentes paires d’atomes et de liaisons ont des distances différentes (par exemple, des liaisons simples et des liaisons doubles). C’est une abstraction très pratique et courante pour décrire cet objet 3D comme un graphique, où les nœuds sont des atomes et les arêtes sont des liaisons covalentes.
Au sein d’un GNN, les nœuds recueillent des informations auprès de leurs voisins, car ils échangent des informations entre eux grâce aux segments qui les relient. C’est ainsi qu’un réseau de neurones en graphe peut apprendre : les informations sont relayées et enregistrées dans les attributs de chaque nœud.
Un des avantages considérables des GNN est leur capacité à tenir compte des relations entre les données (ce qui n’est pas le cas au sein des réseaux de neurones convolutifs). De plus, il est extrêmement difficile d’associer les principes des réseaux de neurones convolutifs sur des données structurées en graphes du fait de la taille arbitraire de ces dernières et de leurs topologies complexes (notamment du fait qu’il n’y a pas de localités spatiales). En effet, le principe de convolution implique de travailler sur des données normées et structurées (par exemple une image) représentées au sein d’un espace géométrique Euclidien. Or, comme vu sur la figure 2, n’importe quelle molécule peut être représentée comme un graphe, et à fortiori les protéines, qui sont des molécules constituées d’un assemblage tridimensionnel d’acides aminés. Cette capacité des GNN à tenir compte des relations entre les données pourrait expliquer, en partie, pourquoi les équipes de Deepmind ont délaissé les CNN et centré leurs efforts sur les GNN dans le cadre du développement de la 2ème version de Alphafold.
3. Les mécanismes d’attention
Les graphes sont des objets mathématiques qui peuvent être manipulés par des mécanismes dits « d’attention ». Ces mécanismes gagnent dorénavant de plus en plus de place dans le champ d’étude et d’expérimentation de l’IA. Les mécanismes d’attention ont été introduits il y a une dizaine d’années et ont récemment permis des avancées considérables dans des domaines tels que le traitement du langage ou de la voix.
Schématiquement, l’attention est une pratique consistant à ajouter une puissance de calcul supplémentaire à certaines données d’entrée. Ceci permet de focaliser le calcul sur un élément (ou un petit nombre d’éléments) des données à traiter. Ces éléments peuvent être des positions dans une image, ou des parties d’un graphe par exemple.
4. Fonctionnement de AlphaFOLD-2
4.1. Les blocs de fonctionnement
Le programme AlphaFold2 est constitué de trois blocs principaux (Figure 3). Le premier bloc est constitué d’une étape de prétraitement de l’information (zone 1 dans la Figure 3). Au sein de ce bloc, la séquence donnée en entrée est utilisée afin de rechercher des informations supplémentaires au sein de bases de données externes. Ces informations sont ensuite intégrées et comparées au sein d’un alignement de séquences multiples (MSA ou Multiple Alignement Sequences) et de représentations par paires. L’ensemble des données est alors envoyé au sein du bloc dénommé « EvoFormer » (zone 2 dans la Figure 3), puis consécutivement transmis au module de structure (zone 3 dans la Figure 3).
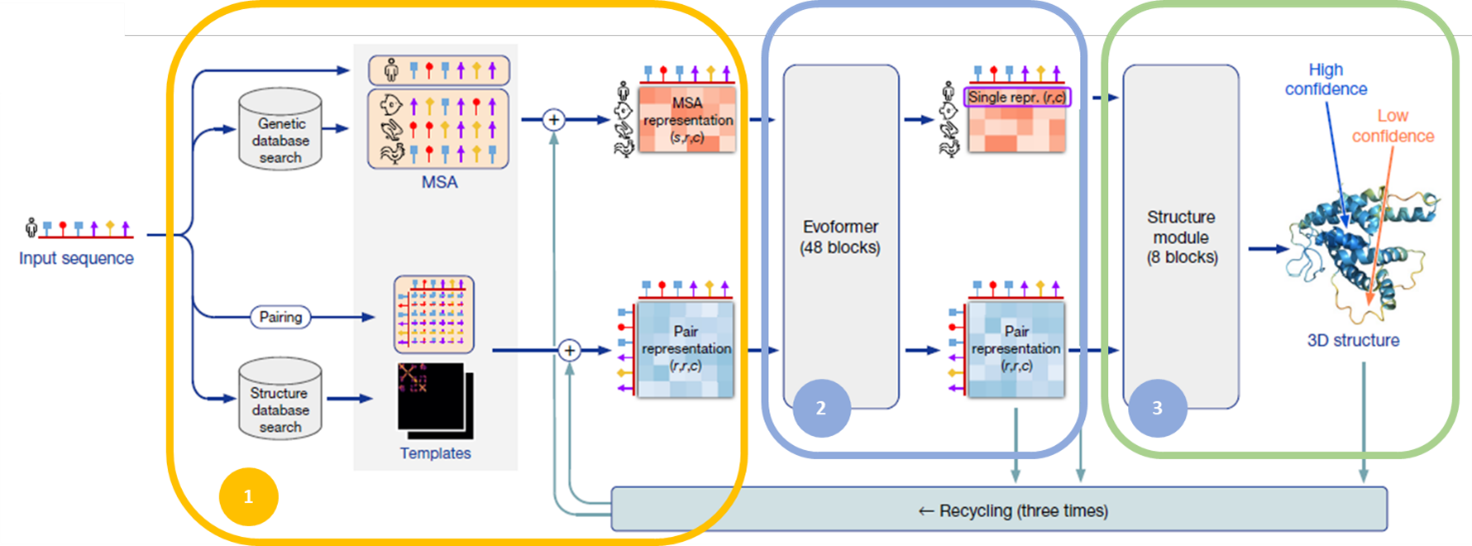
Figure 3 : Représentation du fonctionnement du programme AlphaFold2 de Deepmind. L’architecture de l’IA peut se décomposer en 3 blocs principaux : les données de séquences traitées au sein du bloc d’entrée (1) sont ensuite analysées par le bloc (2) constitués par l’EvoFormer. Les résultats de ces analyses sont ensuite transmis au module de structure (3).
4.2. Le prétraitement des données de séquences
Lors d’une première phase d’apprentissage et sur la base de la séquence d’acides aminés reçue en entrée, AlphaFold2 créé d’abord un alignement de séquences multiples (MSA ou Multiple Alignement Sequences) en cherchant des séquences évolutives similaires, entre différentes espèces notamment, au sein de bases de données de séquences génétiques.
En parallèle, AlphaFold2 cherche également des modèles structurels au sein des bases de données de structures protéiques connues qui possèdent des parties de séquences similaires à celle de la séquence requêtée.
Cependant, durant cette phase d’apprentissage, les modèles structurels analysés par AlphaFold2 peuvent être de mauvaise qualité ou bien ne pas exister au sein des bases de données[8]. Cette disparité dans la qualité des modèles ne permet donc pas à AlphaFold2 de « copier » ces modèles tels quels, mais l’oblige en quelque sorte à réaliser un apprentissage directement à partir des informations de séquences.
En résumé, durant cette première phase d’apprentissage, AlphaFold2 intègre un ensemble d’informations telles que : la séquence d’entrée, le résultat issu des alignements de séquences multiples (recherche d’informations conservées au sein de différentes espèces) (MSA) et des modèles structurels. L’ensemble de ces informations sont regroupées au sein d’un système matricielle de représentation par paires et de MSA[9] (Figure 3 zone 1). L’ensemble des informations issues de cette première phase d’apprentissage est alors utilisé dans la suite du programme.
4.3. EvoFormer
Les données d’alignements précédentes sont ensuite injectées dans le bloc du programme appelé « EvoFormer » (Figure 3 zone 2). L’EvoFormer[10] est constitué de plusieurs blocs de calculs[11] et de deux flux de traitements distincts : un flux dédié au traitement des alignements multiples et un flux dédié aux alignements par paires.
Une des qualités remarquables du fonctionnement de cette partie de l’IA réside notamment dans le fait que le réseau, constitué par le bloc Evoformer, peut itérativement améliorer la qualité des informations issues des alignements multiples (MSA) et des alignements par paires ; tout en permettant aux deux flux contenus au sein du bloc d’échanger des informations. Ceci permet au bloc EvoFormer de travailler directement et simultanément sur les relations spatiales et évolutives dans la protéine. Oui mais comment ?
De fait, en un sens, l’alignement des séquences multiples (MSA) contient des informations sur l’histoire évolutive de la protéine recherchée[12]. Cependant, afin d’extraire l’ensemble des informations pertinentes de ces alignements de séquences multiples, il est également nécessaire d’obtenir des informations sur la structure de la protéine en question ; et c’est justement ce type d’information qui est contenu dans les alignements par paires. D’où le fait que le bloc Evoformer soit structuré de façon à pouvoir utiliser à la fois les données évolutives et structurelles contenues au sein des différents alignements (alignement MSA et alignement par paires).
Au sein de l’EvoFormer, le flux MSA utilise un mécanisme d’attention appelé « attention réelle » par lequel les différentes couches du programme concentrent leur « attention » alternativement sur les lignes et les colonnes des matrices qu’elles reçoivent. L’idée ici étant que l’attention par ligne permet au modèle de raisonner séparément sur les informations de chacune des séquences alignées tandis que l’attention portée sur les colonnes permet au modèle de raisonner sur les relations entre les résidus alignés dans différentes séquences de la pile MSA. Ce mécanisme d’interconnexion des flux de données entre les analyses MSA et les analyses des représentations par paires a lieu dans chacun des 48 blocs qui composent l’EvoFormer, permettant une communication continue entre les représentations MSA et ces représentations par paires.
In fine, chaque bloc constitutif de l’EvoFormer représente les données sous la forme d’un graphique, dans lequel les nœuds sont des paires d’acides aminés et les bords représentent la proximité de ces paires les unes avec les autres au sein de la protéine. Ainsi, chacun des blocs de l’EvoFormer effectue une série de manipulations de ce graphique, en utilisant une variété de techniques d’apprentissage automatique de pointe, avant de transmettre sa prédiction au bloc suivant pour une révision ultérieure. De cette façon, l’ensemble de l’EvoFormer affine progressivement une prévision de ce à quoi devrait ressembler l’épine dorsale de la protéine. Les techniques des mécanismes d’attention utilisées par le système EvoFormer sont similaires à celles qui sous-tendent les récentes percées dans le traitement du langage naturel[13].
4.4. Module de structure
L’ensemble des analyses et des données qui résultent du bloc EvoFormer sont ensuite transférées au module de structure (Figure 3 zone 3). Ce module a pour objectif de transformer les données de séquences d’acides aminés issues du bloc EvoFormer en coordonnées tridimensionnelles afin d’aboutir à une représentation en trois dimensions de la séquence protéique. L’idée remarquable et sous-jacente au module de structure est que, à ce stade, le programme va totalement ignorer les contraintes moléculaires inhérentes à l’enchaînement des acides aminés (positions, angles des molécules les unes par rapport aux autres, espaces pris par chacune des molécules). De fait, lors de la première itération du module de structure, les coordonnées tridimensionnelles de la protéine ne sont pas encore connues et l’ensemble des résidus au sein de la protéine sont modélisés comme étant localisés exactement au même point qui servira d’origine à l’ensemble de l’espace tridimensionnel.
Composé de huit blocs de réseaux neuronaux le module de structure effectue une série de transformations géométriques afin d’affiner davantage la forme probable de la protéine. Pour ce faire, un mécanisme d’attention est également implémenté au sein de ce module afin que celui-ci soit en mesure de calculer la structuration des différentes parties de la protéine prédite indépendemment les unes des autres. Ce module permet ainsi de construire une image des « chaînes latérales » probables de la protéine.
La donnée résultante du bloc de structure se caractérise sous la forme d’une représentation tridimentionnelle de la protéine associée à un degré de confiance quant à l’exactitude des différentes parties qui la composent.
5. Conclusion
Grâce aux progrès incroyables de l’intelligence artificielle, un des grands défis de la biologie moderne est aujourd’hui considéré comme résolu. Comme nous l’avons vu, AlphaFold-2 n’est pas le résultat d’une seule idée révolutionnaire mais plutôt une collection d’idées intelligemment conçues et novatrices qui ont été améliorées au cours de nombreux cycles de conception, ce qui a abouti à ce système remarquable. Cependant, et bien que la prouesse d’’AlphaFold‑2 ouvre la voie à une toute nouvelle ère de la biologie computationnelle assistée par IA, certaines prédictions sont encore hors de sa portée.
Ainsi, AlphaFold-2 ne prédit pas (encore) les structures de complexes formés avec d’autres protéines ou d’autres molécules comme l’ADN et l’ARN, et ne livre pas d’informations sur la dynamique du processus de repliement de la molécule. De plus, la fonction portée par la plupart des protéines est amenée au cours d’une étape de dynamique moléculaire aboutissant à un changement de conformation. De fait, les protéines peuvent changer de conformation au moment où elles s’associent aux cibles sur lesquelles elle se lient. Or, AlphaFold-2 est à l’heure actuelle incapable de modéliser ces changements de conformations essentielles au fonctionnement des protéines car son mode de prédiction est statique. Pour autant, pouvoir modéliser la flexibilité d’une protéine dans ces différentes conformations sera crucial pour étudier les interactions protéine-protéine.
Reste également qu’AlphaFold-2 a été entraîné sur un ensemble de données de protéines qui apparaissent naturellement en biologie et, à l’heure actuelle, il n’est pas possible de savoir à quel point ses prédictions se généraliseront à des protéines complètement nouvelles que nous pourrions vouloir concevoir grâce à de nouveaux outils de design moléculaire.
Enfin, d’autres problèmes inhérents aux réseaux de neurones restent d’actualité avec les GNN, et notamment le phénomène de la « boîte noire » qui peut paraître paradoxale mais qui n’en est pas moins vrai : on ne comprend encore pas bien comment un réseau de neurones, à fortiori un réseau de neurones en graphique, sélectionne son estimation finale car, de l’extérieur, il est presque impossible de suivre (mathématiquement) les processus qui se déroulent au sein du système.
Quoiqu’il en soit, AlphaFold-2 est capable de prédire les structures de la plupart des protéines à domaine unique en biologie avec une précision incroyable et il est indéniable que cela annonce une toute nouvelle ère en biologie computationnelle où l’IA sera utilisée comme un outil indispensable afin de concevoir de nouveaux médicaments, comprendre certaines maladies, ou encore combattre les virus…
[1] Inneance – Lettre Science – Alphafold
[2] Concours CASP (Critical Assesment of Structure Prediction)
[3] https://www.quebecscience.qc.ca
[4] https://www.nature.com/articles/s41586-021-03819-2
[5] (ou CNN pour Convolutional Neural Network)
[6] Un avantage majeur des réseaux convolutifs est l’utilisation d’un poids unique associé aux signaux entrant dans tous les neurones d’un même noyau de convolution. Cette méthode réduit l’empreinte mémoire, améliore les performances et permet une invariance du traitement par translation.
[7] Dans le cas d’un graphe représentant un réseau social, les attributs peuvent être des informations telles que l’identifiant de la personne, le nom, le sexe, sa géolocalisation etc.
[8] En effet, les structures protéiques hébergées au sein de ces bases de données ne sont pas systématiquement de bonne qualité, certaines informations de structures peuvent être manquantes, voir même inexistantes. Ils s’agit très majoritairement de données expérimentales déterminées par cristallographie ou RMN, ce qui explique notamment la mauvaise qualité de certaines annotations protéiques.
[9] En réalité, du fait du grand nombre de données externes disponibles, ce pipeline de prétraitement est répété plusieurs fois, ce qui conduit à différentes versions des alignements par paires et des alignements multiples. Les données issues de ces différentes versions d’alignements sont ensuite moyennées avant d’entrer dans le module de structure.
[10] L’EvoFormer se base sur un modèle d’apprentissage profond : « le transformer ». Les transformers sont conçus pour gérer des données séquentielles (comme le langage naturel) pour des tâches telles que la traduction et la synthèse de texte. L’avantage des transformers réside notamment dans le fait qu’ils n’exigent pas que les données séquentielles soient traitées dans l’ordre. Par exemple, si les données d’entrée sont une phrase en langage naturel, le transformer n’a pas besoin d’en traiter le début avant la fin. Grâce à cette fonctionnalité, le transformer permet une parallélisation beaucoup plus importante que les réseaux de neurones traditionnels et donc des temps d’entraînement réduits. Étant donné que le modèle transformer facilite davantage la parallélisation pendant l’entraînement, il permet un apprentissage sur des ensembles de données plus volumineux qu’il n’était possible avant son introduction (https://arxiv.org/abs/1706.03762).
[11] 48 blocks de calcul exactement.
[12] En effet, les données contenues dans les alignements des séquences multiples (MSA) sont issues de bases de données de séquences génétiques. En un sens, Alphafold2 compare la séquence en entrée à une multitude de séquences issues de différentes espèces. Ce qui explique notamment que les MSA contiennent donc des informations évolutives.

{kind=link}