
Dans les Lettres des Sciences des 13 octobre et 9 juin dernier, Inneance relatait les performances remarquées de l’intelligence artificielle Alphafold développée par la filiale DeepMind de Google, permettant la prédiction de structure 3D de protéines à partir de leurs seules séquences d’acides aminés.
De récents travaux de recherche menés par un consortium de laboratoires et d’instituts de recherche[1] ont combiné des approches par IA avec des techniques expérimentales et des simulations informatiques, afin de mettre en évidence l’architecture 3D du complexe protéique de pore nucléaire humain, de la façon la plus détaillée jusqu’ici.
1. Protéines et pores nucléaires
Pour rappel, les protéines sont des macromolécules constituées d’un assemblage complexe de molécules plus petites : les acides aminés.
Les protéines sont des acteurs essentiels du vivant, responsables d’une grande majorité de l’activité cellulaire. Elles sont capables d’effectuer d’innombrables réactions chimiques complexes et peuvent même interagir entre elles : la vision, la conduction nerveuse, la fabrication de l’énergie chimique cellulaire, la photosynthèse, le déplacement et les interactions cellulaires sont des phénomènes très différents, mais ont pour point commun d’utiliser systématiquement des protéines.
Il existe ainsi une multitude de protéines, chacune ayant une fonction et un rôle spécifiques.
Les pores nucléaires (NPC ou Nuclear Pore Complexe) humains sont de grands complexes protéiques traversant l’enveloppe nucléaire, qui sépare le noyau et son ADN du reste d’une cellule (Figure 1, Figure 2). Ceux-ci sont essentiels au cycle de vie des cellules dans la mesure où ils permettent et contrôlent les échanges, dans les deux sens, entre le noyau et le cytoplasme d’une cellule.
Parmi les activités de régulation des pores nucléaires, on peut citer notamment le blocage de l’entrée des virus dans le noyau, ou encore le transport des ARN messagers (ARNm[2]) du noyau vers le cytoplasme. On sait aujourd’hui que le système de transport nucléaire peut-être impliqué dans plusieurs maladies comme les troubles neurodégénératifs ou certaines infections virales.
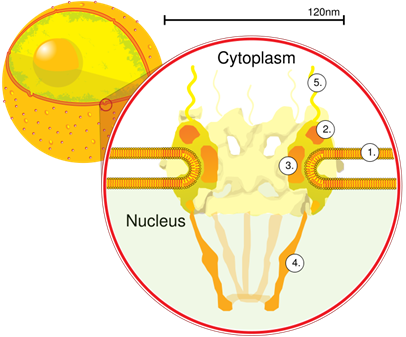
Figure 1 : Représentation schématique d’un pore nucléaire (en coupe). La structure d’un pore nucléaire peut se décomposer en quelques éléments. 1. Enveloppe nucléaire. 2. Anneau externe. 3. Anneau interne. 4. Panier. 5. Filaments. Source
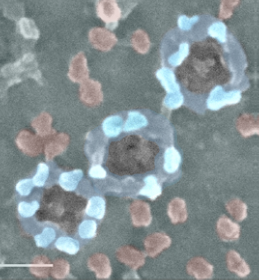
Figure 2 : Cliché de pores nucléaires en microscopie électronique à balayage (Walther et al., 2002). Vue de la face cytoplasmique de l’enveloppe nucléaire. NB : les protéines du pore sont colorées en bleu. La taille de la barre blanche équivaut à une distance de 100 nm sur l’image.
Le pore nucléaire est le plus grand complexe protéique de la cellule : il est constitué d’un assemblage d’environ 1000 protéines, et environ 100 protéines de types différents entrent dans sa composition. Loin de constituer un enchevêtrement protéique statique, l’architecture des pores nucléaires est très dynamique. De fait, les pores nucléaires peuvent répondre aux variations de tension pouvant se manifester au niveau de l’enveloppe nucléaire par un changement conformationnel qui se manifeste, par exemple, par des mouvements de dilatation et de constriction de son anneau central.
Jusqu’à présent, les modèles structuraux des pores nucléaires mis en avant par la communauté scientifique couvraient moins de 50% de sa structure et présentaient systématiquement un manque de précision dû au fait que beaucoup de structures ont notamment été résolues par comparaison d’espèces relativement éloignées de l’espèce humaine. Par « résoudre une structure », on entend élucider sa conformation spatiale.
De plus, la localisation transmembranaire du complexe protéique rend son élucidation difficile. Pour autant, la clarification de son architecture permettrait d’acquérir une meilleure compréhension de la fonction et du dynamisme des pores nucléaires.
2. Intelligence artificielle et approche expérimentale
De récents travaux de recherche, publiés au sein de la revue Science, ont permis de créer le modèle de pore nucléaire le plus complet à ce jour. Cette avancée a notamment été permise par une approche utilisant le programme de prédiction de la structure des protéines Alphafold2 de DeepMind combinée à des techniques telles que la cryo-tomographie électronique ainsi que la modélisation intégrative. Grâce à la combinaison de ces approches expérimentales et informatiques, le modèle 3D de pore nucléaire révélé par les scientifiques couvre plus de 90% de la structure du complexe protéique, soit la meilleure précision obtenue à ce jour.
Dans un premier temps, les chercheurs ont notamment utilisé la cryo-tomographie électronique afin de modéliser globalement le complexe protéique. Cette technique combinant microscopie et tomographie à basse température permet d’obtenir la restitution tridimensionnelle d’un objet à partir d’images bidimensionnelles. Grâce à cette technique, les chercheurs ont pu observer le pore nucléaire dans son environnement cellulaire, plutôt qu’isolé. Par la suite, l’utilisation d’AlphaFold2 a permis de révéler plus de détails sur les éléments constitutifs des protéines individuelles du complexe (Figure 3).
Puis les chercheurs ont eu recours à ColabFold, une version modifiée d’AlphaFold2 permettant de modéliser les interactions entre protéines. Le recours à cette IA leur a ainsi permis de visualiser les différents sous-complexes protéiques constituant le pore nucléaire.
Figure 3 : Vue en coupe de la modélisation du pore nucléaire. Les différents sous-complexes protéiques sont représentés par différentes couleurs.
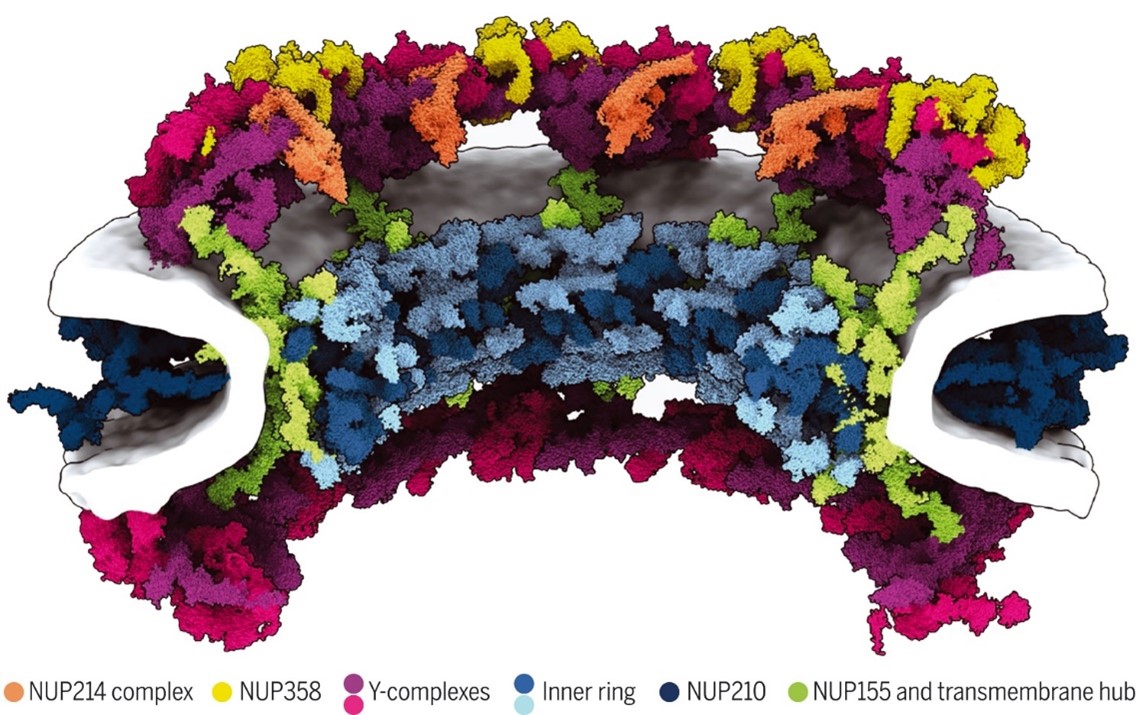
Figure 4: Comparaison de la structure connue du complexe de pores nucléaires en 2016 (à gauche) et 2022 (à droite). Les complexes protéiques nouvellement élucidés sont représentés en orange et en jaune. Source

3. Résultats
Le modèle issu de ces travaux de recherche couvre plus de 90% de la structure du pore nucléaire (Figure 4). Cette modélisation a notamment permis aux chercheurs d’observer les changements conformationnels se produisant au cours de la contraction et de la dilatation du complexe et a également révélé la localisation de différents sites d’ancrage. Par ailleurs, la structure révélée par les chercheurs leur a également permis de comprendre comment les protéines du complexe et la membrane nucléaire interagissent afin de former un pore stable et dynamique. Ils ont également pu appréhender la manière dont celui-ci répond aux signaux mécaniques tels que la dilatation et la constriction de la membrane nucléaire.
Comme évoqué précédemment les pores nucléaires assurent un rôle central dans la coordination du transport de différentes molécules, ainsi que la transcription et la maturation des ARNm ; ce qui en fait un site sensible aux mutations pouvant provoquer différentes maladies ou aux interactions hôtes‑pathogènes. Les découvertes associées à la compréhension de sa structure sont par conséquent des éléments essentiels de la recherche en thérapie génique, les vaccins de type ARNm, ou encore les technologies CRISPR[3].
[1] Le groupe Kosinski de l’EMBL de Hambourg et du Centre de biologie des systèmes structurels (CSSB), des laboratoires Beck et Hummer de l’Institut Max Planck de biophysique
[2] L’ARNm est une molécule faisant office de copie transitoire entre les gènes codés au sein de l’ADN dans le noyau de la cellule, et la synthèse de protéine qui a lieu dans le cytoplasme.
[3] En génie génétique, le système CRISPR-Cas9, est un outil de manipulation génétique notamment utilisé comme « ciseau moléculaire » afin d’introduire des modifications locales du génome (on parle aussi d’édition génomique) de nombreux organismes modèles.
