L’intelligence artificielle Alphafold développée par la filiale DeepMind de Google, a remporté le dernier concours CASP[1] de prédiction de structure 3D de protéines à partir de leurs seules séquences d’acides aminés. Il s’agit de la première IA à prédire la structure de protéines avec une précision quasiment égale à celle atteinte lors d’expériences en laboratoire, et en nettement moins de temps.
Depuis la fin des années 1960, les chercheurs en biologie s’intéressent au phénomène de repliement des protéines, ces grandes molécules dont la longue chaîne d’acides aminés s’agence de façon à adopter une structure en 3D qui leur est propre et leur confère leurs fonctions. Cependant, déterminer leur structure tridimensionnelle expérimentalement est un processus laborieux et coûteux, et la communauté scientifique tentait jusqu’ici, sans succès, de mettre en place des techniques de bio-informatique permettant de prédire de tels agencements, à partir de leur seule séquence polypeptidiques (séquences linaire d’acides aminés).
Récemment, un article[2] publié dans la prestigieuse revue Nature annonçait que l’intelligence artificielle Alphafold développée par Deepmind avait réalisé « un pas de géant » en matière de détermination de structures protéiques : déterminer la structure de protéines en 3 dimensions à partir de leurs seules séquences d’acides aminés.
De fait, l’IA mise au point par la firme de Google a largement surpassé tous ses concurrents lors de la quatorzième édition du CASP, une compétition biannuelle qui réunit les spécialistes de la biochimie structurale théorique. Ayant atteint un niveau de précision jusque-là inégalé dans le domaine, AlphaFold pourrait complètement changer la donne, et révolutionner la bio-ingénierie et la recherche au sens large.
Protéines : des molécules essentielles à la vie
Les protéines sont des macromolécules constituées d’un assemblage complexe de molécules plus petites : les acides aminés. Les protéines sont des acteurs essentiels du vivant, responsables d’une grande majorité de l’activité cellulaire. Celles-ci sont capables d’effectuer d’innombrables réactions chimiques complexes et peuvent même interagir entre elles : la vision, la conduction nerveuse, la fabrication de l’énergie chimique cellulaire, la photosynthèse, le déplacement et les interactions cellulaires sont des phénomènes très différents, mais tous utilisent des protéines. Il existe ainsi une multitude de protéines, chacune possédant une fonction et un rôle spécifiques. À titre d’exemple, ce sont des protéines qui catalysent les réactions chimiques de synthèse et de dégradation nécessaires au métabolisme de la cellule, qui assurent un rôle structurel au sein du cytosquelette ou des tissus (actine, collagène). Certaines protéines peuvent être vues comme des « moteurs moléculaires » qui permettent la mobilité (myosine), d’autres sont impliquées dans le repliement de l’ADN (histones), la régulation de l’expression génétique (facteurs de transcription), le métabolisme énergétique (ATP synthase) ou encore la transmission de signaux cellulaires (récepteurs membranaires).
Dans le cadre du repliement des protéines, il y a une notion essentielle à prendre en compte, qui est que la structure détermine la fonction : le rôle d’une protéine donnée dépend entièrement de sa structure tridimensionnelle (ou conformation). Ainsi, lors du repliement d’une protéine, la séquence d’acides aminés se replie sur elle-même, adoptant une configuration particulière. Cette configuration est fonction des interactions physico-chimique existantes entre chacun des acides aminés. Au cours de ce repliement, certains acides aminés vont ainsi se rapprocher les uns des autres et pouvoir notamment former une (ou des) unités fonctionnelles, tandis que d’autres vont se repousser et seront par exemple maintenus en périphérie de la molécule. L’enchaînement « linéaire » des acides aminés est donc prépondérant et détermine la forme et la fonction des protéines.
L’étude des protéines : un enjeu de santé
Élucider les mécanismes de repliement des protéines est l’un des enjeux majeurs de la biologie moderne. En effet, l’étude du repliement des protéines a de multiples applications pratiques en médecine, en permettant notamment la compréhension des différentes pathologies associées aux repliements incorrects telles que les maladies de type amylose (comme la maladie d’Alzheimer), la maladie de Creutzfeldt-Jakob et de nombreuses autres formes de protéinopathie comme la fibrose kystique. Par ailleurs, l’étude et la compréhension des mécanismes de repliement des protéines peut permettre la conception et le développement de nouvelles protéines (enzymes de synthèse et nouvelles fonctions associées), ou de nouveaux médicaments.
Déterminer la structure des protéines
Pendant des décennies, les expériences en laboratoire ont été les principaux moyens de déterminer la structure des protéines. Les premières structures complètes ont été déterminées à la fin des années 1950, grâce à une technique qui consiste à envoyer des faisceaux de rayons X sur des protéines cristallisées[3]. L’utilisation de la cristallographie aux rayons X a ainsi permis d’obtenir une grande partie des structures protéiques connues. Puis, la spectroscopie par résonance magnétique nucléaire (RMN), a également permis d’exploiter les propriétés magnétiques de certains noyaux atomiques[4]. Un peu plus tard est apparu la cryomicroscopie électronique[5]. Cependant, ces méthodes présentent l’inconvénient majeures d’être expérimentales, fastidieuses, longues à mettre en place, très coûteuses, et non sans défauts[6].
Pour autant, il est théoriquement possible de prédire la configuration tridimensionnelle d’une protéine sur la base de la connaissance de sa seule séquence linéaire d’acides aminés, à la condition de savoir comment les différents éléments interagissent entre eux. De fait, un programme informatique devrait être capable de modéliser un tel repliement, en testant un ensemble de combinaisons possibles jusqu’à obtenir celle qui sera optimale pour la protéine donnée.
De fait, les premières tentatives d’utilisation des ordinateurs pour prédire la structure des protéines dans les années 1980 et 1990 ont donné de piètres résultats. Des méthodes proposées pour certaines protéines se révélaient inopérantes lorsqu’elles étaient appliquées à d’autres protéines.
De plus, un obstacle majeur a mis un frein au développement de ce genre de techniques de bio-informatique : même si, dans la nature, les protéines se replient en à peine quelques millisecondes, le nombre de façons dont elles pourraient théoriquement se replier avant de se fixer dans leur structure 3D finale est simplement astronomique. Déjà en 1969, le biologiste moléculaire américain Cyrus Levinthal estimait qu’énumérer toutes les configurations possibles d’une protéine typique prendrait plus longtemps que l’âge actuel de l’Univers[7].
Cependant, au cours de la deuxième moitié du XXème siècle, les progrès réalisés en termes de composants électronique et de techniques informatiques ont permis d’augmenter les capacités de calcul et d’apprentissage des machines, ainsi que leur vitesse d’intégration des données et d’exécution des tâches.
CASP et Alphafold
En fin d’année dernière s’est tenue la conférence du 14ème concours CASP, au cours duquel des équipes internationales sont en compétition et cherchent à prédire les structures de protéines qui ont déjà été résolues expérimentalement mais qui n’ont jamais été publiées. Le concours CASP évalue ainsi les modèles de prédiction des différents concurrents en mesurant les différences entre la structure protéique prédite et la structure réelle. Durant plusieurs mois, les participants reçoivent des séquences régulièrement (près d’une centaine au total) et ont à chaque fois quelques semaines pour rendre leurs prédictions structurelles. Une équipe de chercheurs indépendante évalue ensuite les prédictions à l’aide de paramètres qui mesurent la similarité entre la structure prédite et la structure déterminée expérimentalement. Cette évaluation est anonyme : les examinateurs ne savent pas quelle équipe est derrière chaque prédiction.
La principale métrique permettant de mesurer la précision des prédictions est le Global Distance Test (GDT). Ce test permet de déterminer un score afin d’évaluer chaque prédiction de structure de protéine. Le GDT peut être approximativement considéré comme le pourcentage de résidus d’acides aminés positionnés à une distance seuil de la position correcte (déterminée expérimentalement). Un score de 90 GDT ou plus est considéré comme égal à une analyse expérimentale. Au cours du CASP 14, le score médian d’AlphaFold par rapport à toutes les protéines cibles prédites était de 92,4 GDT. Cela traduit notamment le fait que les prédictions réalisées par AlphaFold ont une précision telle que l’erreur moyenne des prédictions est d’environ 1,6 angström[8], soit à peu près la largeur d’un atome (Figure 1).
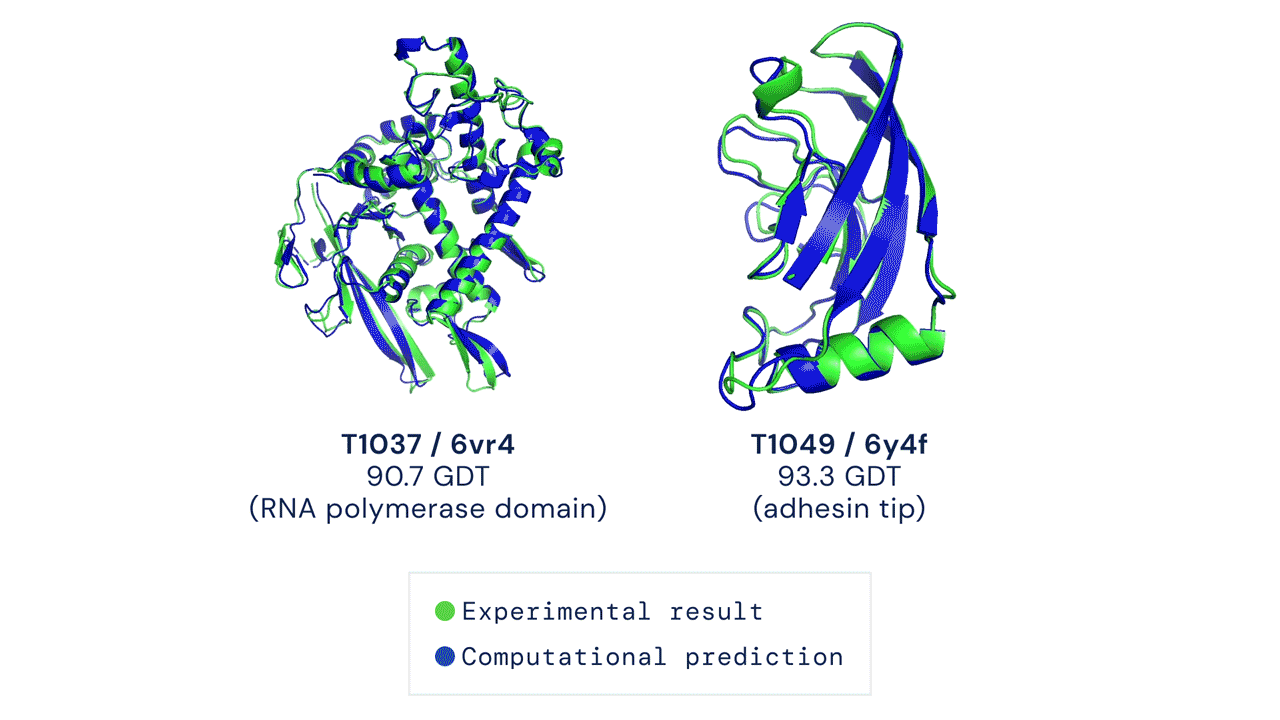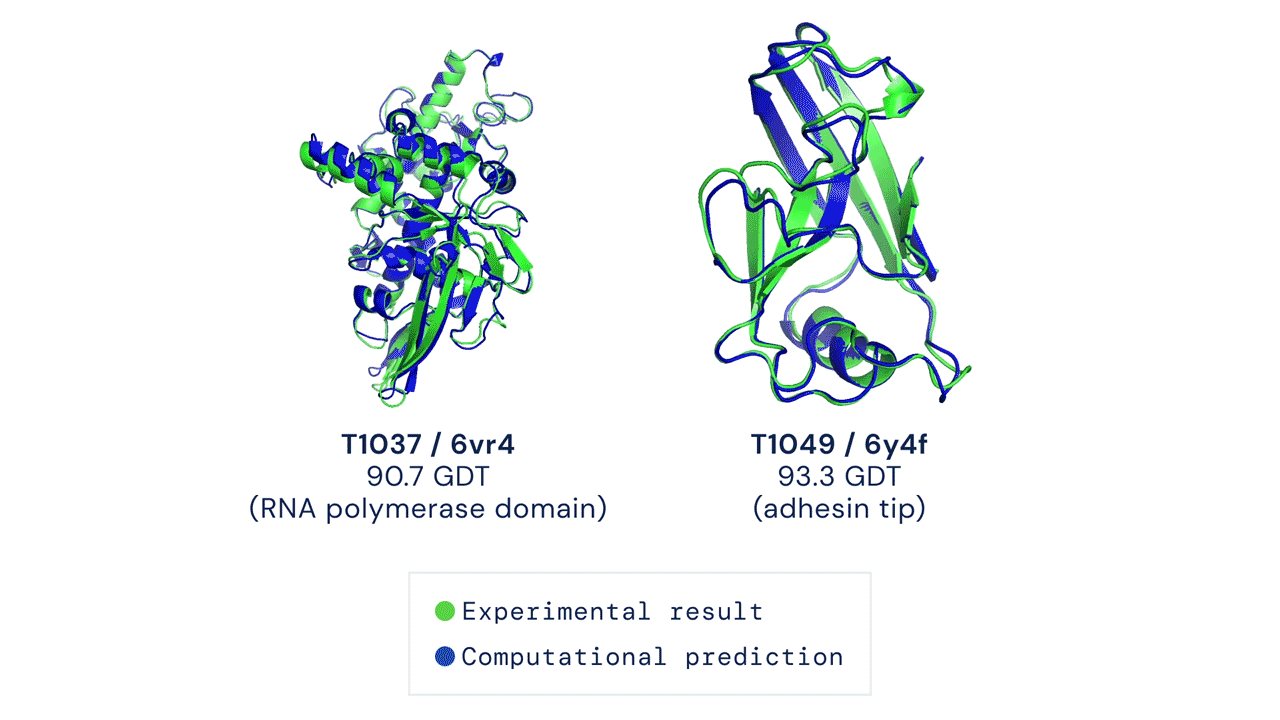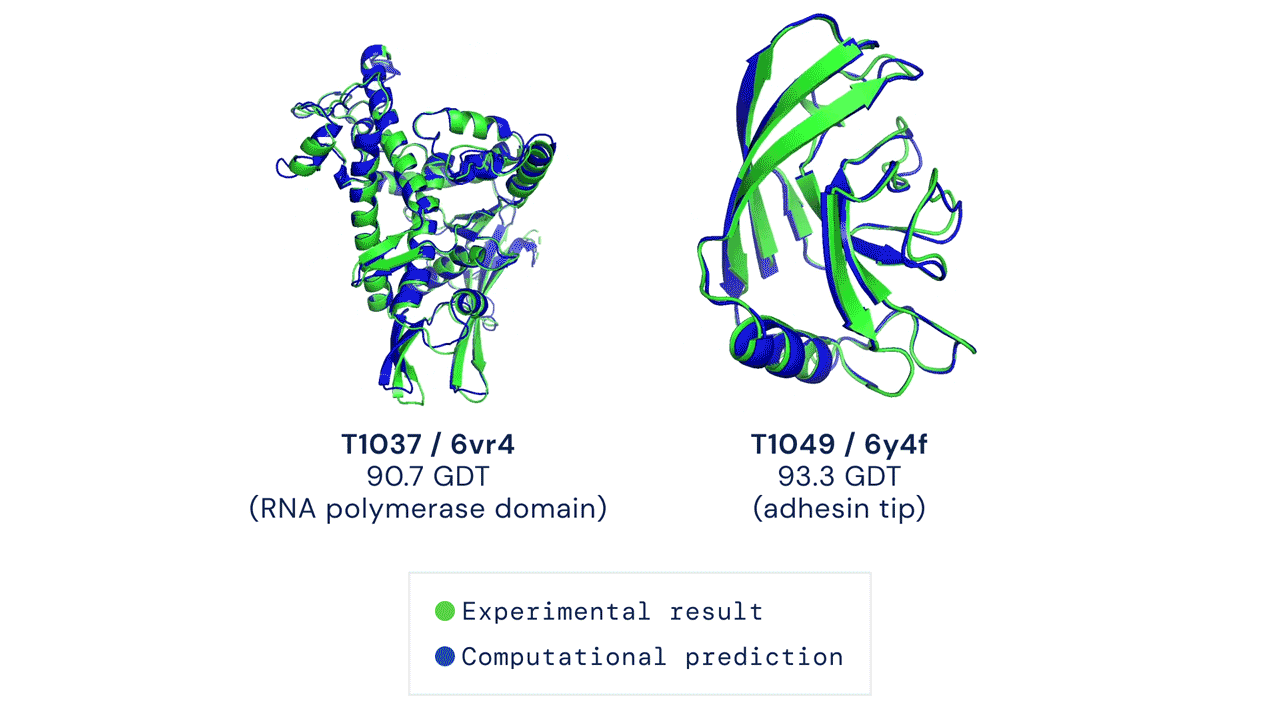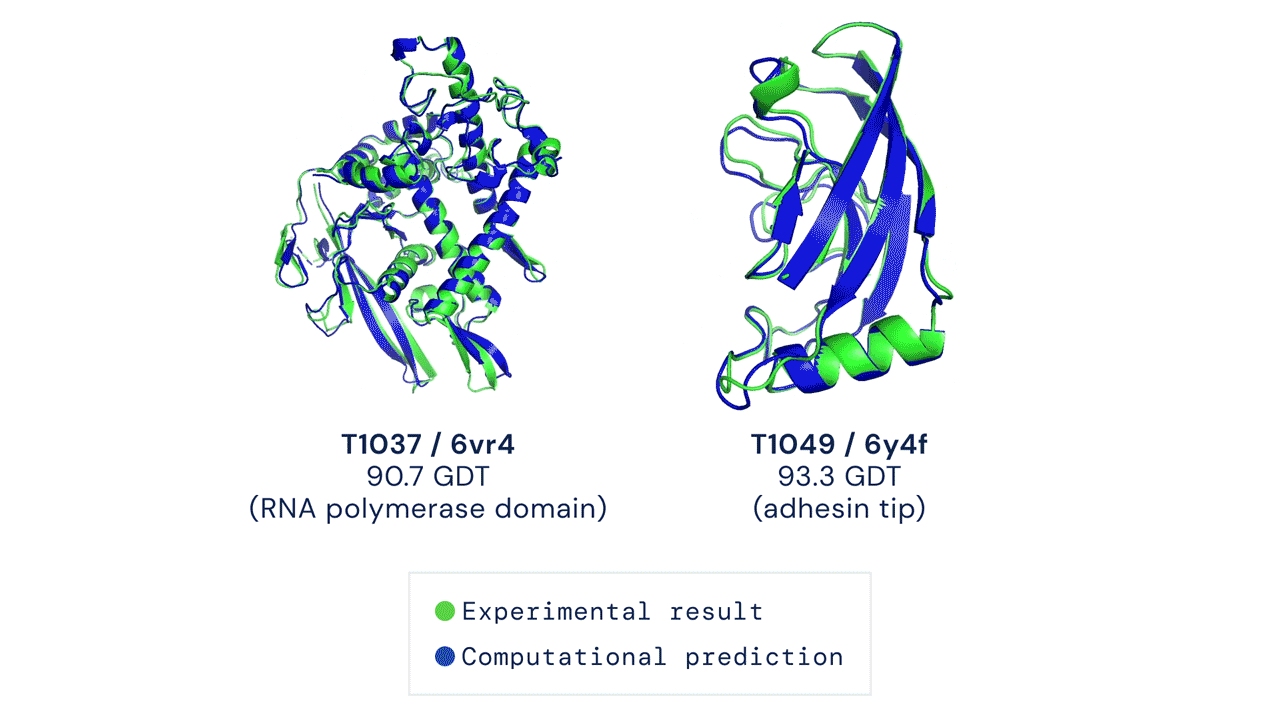
Figure 1 : La figure montre deux prédictions de structures protéiques réalisé par AlphaFold. Les données expérimentales ainsi que les données prédites sont superposées à titre d’illustration. En vert, les données structurales obtenues par analyse expérimentale. En bleu, la conformation prédite par AlphaFold.
Une des structures prédites a notamment permis de résoudre la conformation d’une protéine bactérienne impliquée dans des phénomènes de résistance aux antibiotiques et sur laquelle la communauté scientifique butait depuis près d’une décennie. Cette performance a été réalisée par AlphaFold en seulement 30 minutes avec un degré de précision de 92%[9].
Cette performance est d’autant plus intéressante que l’on sait que AlphaFold s’est entraîné sur un jeu de données relativement modeste, avec seulement quelques 170 000 structures de protéines connues dans les bases de données publiques.
Pour autant, et bien que cette réussite soit tout à fait remarquable, certaines problématiques restent à l’heure actuelle encore difficiles à résoudre. Ainsi, à l’heure actuelle, AlphaFold ne peut pas prédire les structures de complexes formés avec d’autres protéines (complexe protéique) ou d’autres molécules comme l’ADN et l’ARN, et ne livre pas non plus d’information sur la dynamique du processus de repliement de la molécule.
Mais il est certain que l’IA et l’apprentissage profond vont considérablement accélérer la détermination des structures protéiques ce qui s’avèrera un atout indéniable à différents niveaux comme par exemple, permettre de formuler des hypothèses sur les fonctions biologiques des protéines prédites, ou bien tester ces protéines in silico (en introduisant par exemple des mutations le long des chaînes d’acides aminés), en vue de traiter certaines maladies ou de neutraliser des agents pathogènes.
Tout récemment, Deepmind a publié et rendu accessible publiquement le programme source d’AlphaFold. De plus, le LEBM (Laboratoire Européen de Biologie Moléculaire), s’est associé à DeepMind pour mettre en place une base de données en accès libre, qui contient déjà plus de 350 000 structures tridimensionnelles de protéines prédites à l’aide de l’IA – et probablement plusieurs millions d’ici à quelques mois.
L’ensemble de ces avancées vont permettre l’émergence d’une nouvelle ère de la biologie moléculaire. Les questions posées ainsi que les réflexions scientifiques abordées seront plus complexes, tandis que les manipulations et l’expérimentation telles qu’elles se définissent encore aujourd’hui tendront à diminuer.
[1] Concours CASP (Critical Assesment of Structure Prediction) : https://predictioncenter.org/
[2] https://www.nature.com/articles/d41586-020-03348-4
[3] La cristallographie aux rayons-X est une technique utilisée en laboratoire permettant d’obtenir des informations structurelles de molécules. La cristallographie aux rayon X emploie le rayonnement électromagnétique (rayon X). La lumière est diffractée par les électrons de la molécule, et l’analyse du motif de diffraction obtenu permet la localisation des atomes de la protéine (cristallisée), et sa reconstitution tridimensionnelle.
[4] Une analyse RMN protéique suit habituellement les étapes suivantes : préparation de l’échantillon, acquisition et traitement du signal RMN produit par les noyaux observés, analyse et attribution des spectres, et enfin calcul des coordonnées des atomes à partir des paramètres mesurés sur les spectres RMN. L’analyse RMN est une technique qui exploite les propriétés magnétiques de certains noyaux atomiques en se basant sur le phénomène de résonance magnétique nucléaire (RMN).
[5] Cette technique consiste à prendre un grand nombre d’images en microscopie électronique d’un échantillon cryogénisé et, par un traitement statistique, à en déduire des images moyennes de la molécule étudiée vue sous différents angles pour pouvoir la reconstruire en 3D
[6] Dans le cas de la cristallographie aux rayons X, l’échantillon analysé doit être cristallisable et les types d’échantillons, dont l’analyse est possible, restent limités (il est par exemple difficile de cristalliser des protéines de membranes). Dans le cadre de la RMN, l’obtention d’une structure tridimensionnelle de la protéine étudiée est conditionnée par les choix qui sont réalisés en amont de l’étude proprement dite. Ces choix concernent notamment la définition précise du ou des polypeptides à étudier, la stratégie d’attribution des signaux RMN, ainsi que la méthode de production de l’échantillon. Dans le cadre de la cryomicroscopie, la préparation des échantillons est complexe et l’appareillage extrêmement coûteux.
[7] Levinthal C. Are there pathways for protein folding ? J Chem Phys 1968; 65 : 44–5.
[8] Un angström ou est une unité de longueur utilisée pour mesurer les distances très petites. 1 angström est égale à 10 -10 mètres et correspond au diamètre d’un atome.
